

"Periodic Table" of proteins helps make sense of structure
BERKELEY – Scientists at the University of California, Berkeley, and Lawrence Berkeley National Laboratory have taken the first stab at a "periodic table" of the protein structures - an organized map of the building blocks used over and over again to construct the billions of complex proteins that make up life on Earth.
 The protein from the cancer-causing oncogene called ras provides one example of a protein's complex three-dimensional geometry. (Credit: Sung-Hou Kim/UC Berkeley) |
The three-dimensional map depicts similarities and differences among the building blocks, letting scientists visualize the universe of possible protein structures - the many possible twists, turns and folds - and see evolutionary changes that may have occurred with time. The map may help them understand the relationship among all proteins in nature in the same way that the periodic table of the elements gave chemists a framework for understanding how molecules are related.
"This is a very good way to organize and visualize the whole protein universe," said Sung-Hou Kim, professor of chemistry at UC Berkeley and head of the Structural Biology Department of the Physical Biosciences Division at Lawrence Berkeley National Laboratory. "The major impact of this research will be conceptual, providing a global view of protein structure and how different structures may have evolved."
Since understanding the molecular functions of proteins is key to understanding cellular functions, the map holds promise for a number of areas of biology and biomedical research, including the design of more effective pharmaceutical drugs that have fewer side-effects, Kim said.
"This map can be used to help design a drug to act on a specific protein and to identify which other proteins with similar structures might also be affected by the drug," he said.
Kim and his colleagues, UC Berkeley graduate students Jingtong Hou and Gregory Sims, and research associate Chao Zhang, published their map this week in the online early edition of the Proceedings of the National Academy of Sciences (PNAS). The article will be posted on the PNAS Web site sometime between Feb. 18 and 21.
Kim is a member of UC Berkeley's Health Sciences Initiative, a campus-wide collaboration that brings the physical sciences together with the biological sciences to tackle health issues of the 21st century. He also is a member of the California Institute for Quantitative Biomedical Research (QB3), a cooperative effort among three UC campuses and private industry that harnesses the quantitative sciences to integrate our understanding of biological systems at all levels of complexity.
As the number of organisms with completely sequenced genomes passes 100, with another 600 expected by the end of the year, scientists are awash in gene sequences for proteins. The human genome alone has nearly 40,000 genes.
Yet, gene sequences are only the starting point in understanding an organism. The unique protein for which each gene codes is the real workhorse. Proteins are both the house and the housekeeper, the function of each determined by its three-dimensional structure and how it networks with other proteins. To date, however, scientists know the structure - the folds and twists - of only about 20,000 proteins from a broad variety of organisms. Among all organisms on Earth, there could be as many as a trillion different proteins.
Many once thought that, with enough computing power, scientists eventually would be able to predict protein structure from the amino acid sequence alone. That goal is very far from being accomplished, if it is possible at all, Kim said.
As a protein structure chemist, Kim explored several prediction schemes, but abandoned them after realizing that proteins with as little as 10 percent identity in sequence can have nearly the same structure. Instead, he began looking for a global view of the protein universe that would at least let him rule out some predicted structures and search only within the realm of structures nature has favored.
Others, too, have looked for an organizing principle, cataloging types of folds and twists - structural domains or motifs, Kim calls them - common to most proteins. But none of the previous results was understandable from an evolutionary point of view.
His group's method involved mathematically comparing each motif to every other motif, calculating their dissimilarity as a measure of structural "distance" between a given motif pair. The less similar, the greater a pair's structural distance. Then, the group found an arrangement in space such that the physical distance between motifs corresponded to their structural "distance." In the resulting map, the folds or motifs most like one another, the ones that are structurally related, end up plotted next to one another.
Though structural biologists predict there may be 10,000 or more different motifs or building blocks, only about 1,000 are currently recognized from the 20,000 known protein structures. Because the computation involved was very intensive, Kim's group chose a subset of these to "map" - the 500 most common motifs that are the building blocks of about 80 percent of all known protein structures.
"My guess is that three-quarters of most organisms' proteins have structural motifs among these 500," he said. Motifs represent significant differences in folding. An alpha helix is one common structural element of protein motifs, consisting of a helical spiral of amino acids, like a right-handed curled ribbon. For a motif with two helices, two parallel helices represent a motif distinct from two anti-parallel helices.
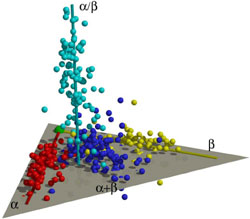 A three-dimensional plot of nearly 500 building blocks or motifs found naturally in folded proteins, showing clustering that appears to be related to the evolution of complex folds. Each sphere represents a protein family exhibiting similar folds. Protein folds that belong to the alpha-helix, beta-sheet and alpha/beta class are mostly clustered around three separate axes, (red, yellow and cyan, respectively). Most protein folds of the alpha+beta class (blue), which are a random mixture of alpha-helices and beta-sheets, fall within a plane between the alpha and beta axes. The alpha- and beta-axes approximately intersect at a single point indicated by the green ball. (Credit: Sung-Hou Kim/UC Berkeley) |
Working with 498 different motifs was computationally intensive - Kim's group occupied all of the lab's computers for more than a week for one set of calculations - but it provided a 3-D map of the protein universe that surprised even Kim. Essentially, the different motifs cluster in four groups that, when plotted in three dimensions, resemble three cigars tied together at one end, with a fourth cigar jutting out from the plane of the other three. The area where the three cigars intersect contain the simple folds, while the more complex folds are found toward the ends of the cigars.
"I didn't expect this," he said. "Other studies showed three clusters of proteins, each with different structural elements, but in our case, we see four clearly segregated groups, and each group has an elongated distribution that shows how it may have evolved.
"The distribution is not random. Only certain classes evolved. It looks like architectural constraints of structure played a restrictive role in early evolution."
One evolutionary pathway elaborated on the alpha helix structure. Another developed from the basic beta sheet, a pair of side-by-side chains of amino acids stabilized by bonds along their length. A third pathway is essentially a random mix of alpha helices and beta sheets.
The fourth path, which diverges from the third about one-third of the way along its cluster, seems to have evolved later. It represents a specific mix of one alpha helix and two beta sheets that has been used as a convenient module to build larger proteins.
"If the other motifs are composed of bricks, this fourth group of motifs is composed of prefabricated units of multiple bricks used to build proteins," Kim said.
To see if the apparent evolutionary relationships suggested by the map are real, Kim and his group compared plots of the proteins from three individual organisms. A salt-loving microbe, Halobacterium, from the primitive group called Archea, contained proteins with more of the simpler structural motifs, while a more evolved bacterium, Chlamydia, had more complex motifs in its proteins. Among bacteria, one of the earliest, the hot springs dweller Aquifex, didn't have as many complex modules as Chlamydia, a more evolved bacterium.
"The map seems to have a loose evolutionary clock embedded in it," Kim said. "It shows the alpha helices and beta sheets developing early, then they started mixing, and finally, one of the mixes took off."
In terms of predicting the structure of unknown proteins, the global protein map Kim's group has constructed shows the types of folds likely to exist in proteins, and eliminates many possible alternatives that nature never used.
Kim's next goal is to add to the map the less common folding motifs or building blocks. In fact, his group has already added many more folds and has found that they occupy the same "envelope" of the four elongated clusters in fold space, suggesting that this "map" constructed of the 498 most common folds is a good representation for all proteins. The Berkeley Structural Genome Center, which he directs, was established two and a half years ago along with eight similar centers in the United States to find the other 9,000 or so structural motifs or families in the protein structure universe. These centers are funded by the National Institutes of Health's Protein Structure Initiative.
He also wants to look at the protein fold maps of the many individual organisms whose genomes have been sequenced to see the differences of distribution of the protein folds.
"This is just the beginning, a first glimpse of the distribution of proteins. There is still a lot of mining and digging to do," Kim said. "This is such a new way of conceptualizing the protein universe, some people have difficulty appreciating it. But others are fascinated by this global view of the protein universe."
Funding for the project came from the National Science Foundation and from the National Institutes of Health through the Berkeley Structural Genome Center.