

'Evolved' virus may improve gene therapy for cystic fibrosis
| 17 February 2009
BERKELEY — Researchers from the University of California, Berkeley, and the University of Iowa have turned a relatively benign virus into a highly infectious form that is ideal as a carrier for gene therapy.
In its first gene therapy test, it completely cured human cystic fibrosis lung tissue in culture.
 UC Berkeley researchers forced the adeno-associated virus to evolve so as to bind to sialic acid on the surface of lung cells, making it easier for the virus to infect them. The forced mutation (red) surrounds the mouth (green) of the receptor that binds sialic acid. (Schaffer lab/UC Berkeley)
UC Berkeley researchers forced the adeno-associated virus to evolve so as to bind to sialic acid on the surface of lung cells, making it easier for the virus to infect them. The forced mutation (red) surrounds the mouth (green) of the receptor that binds sialic acid. (Schaffer lab/UC Berkeley)"I think it is worthwhile thinking about clinical therapy at the levels of infection we are achieving," said coauthor David Schaffer, professor of chemical engineering at UC Berkeley.
A new pig model of cystic fibrosis developed last year by Schaffer's colleague, pulmonologist Joseph Zabner of the University of Iowa Hospitals and Clinics in Iowa City, will provide a key test of the virus as a carrier of a gene to replace the mutated gene responsible for the disease.
"If we are able to show that efficient gene transfer can result in gene therapy, if we can cure the lung disease of pigs that have been genetically engineered to have cystic fibrosis lung disease, we should have a real chance of curing cystic fibrosis in humans," Zabner said in an e-mail.
Schaffer's lab is collaborating with groups elsewhere to adapt the virus to gene therapy for other diseases, including Alzheimer's disease and amyotrophic lateral sclerosis (Lou Gehrig's disease).
"Both of those are situations where improvements in the properties of the vehicle can have a significant impact on the success of the therapy,"
Schaffer said.
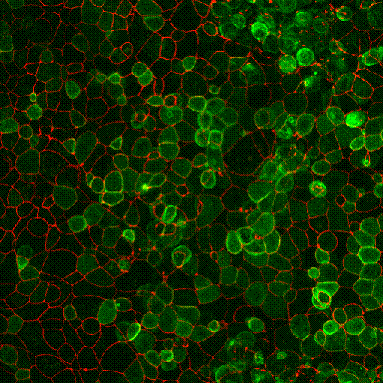 These lung cells from a cystic fibrosis patient have been infected with the evolved virus carrying a correct copy of the CFTR gene. The cells with a green interior are expressing high levels of normal CFTR protein, which is a chloride ion transporter that is defective in CF patients because of a mutation in the CFTR gene. The chloride ion transport of these cells was completely repaired with the virus. (Schaffer lab/UC Berkeley)
These lung cells from a cystic fibrosis patient have been infected with the evolved virus carrying a correct copy of the CFTR gene. The cells with a green interior are expressing high levels of normal CFTR protein, which is a chloride ion transporter that is defective in CF patients because of a mutation in the CFTR gene. The chloride ion transport of these cells was completely repaired with the virus. (Schaffer lab/UC Berkeley)Schaffer and his UC Berkeley colleagues collaborated with Zabner's laboratory to test a technique developed by Schaffer to force the evolution of a virus in ways that make it more effective in gene therapy.
Two years ago, Schaffer and colleagues used the technique to create a variant of AAV that more easily avoids the immune system, allowing the virus to remain in the body long enough to deliver a gene to its intended target.
Using the same technique, the team produced a variant of AAV that is several hundred times more effective at entering lung cells than the natural virus.
Schaffer's technique involves making many mutations in AAV, culturing these variants with cells, and then taking those with specific improved properties
- in this case, the ability to infect lung cells - and repeating the process.
"We probably conducted about six rounds of evolution in which we infected the lung epithelial cultures in Iowa, they sent it back to us, we recovered the viral sequences, made new viruses and sent them back again," Schaffer said. "It was iterative rounds of infection and selection for improved infection that finally led to this enhancement of function."
The main problem in CF is a mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene that results in a defective chloride ion channel in the body's cells. This, in turn, creates a chloride ion imbalance in the cell, which interferes with water transport in and out of the cell. In the lungs, this causes the mucus that lines the lung surface to become thick and sticky. Breathing becomes difficult if the mucus is not loosened, often by vigorous pounding on the chest, and coughed out.
Respiratory infections are common, and lung failure often results. The ion channel defect also affects digestion, leading to nutritional deficiency.
According to Schaffer, previous attempts to deliver a normal CFTR gene to lung cells by means of a virus failed either because the immune system mopped up the virus before it had a chance to deliver its cargo, as was the case with adenovirus; or because the virus was inefficient at delivering the gene to cells, the case with adeno-associated virus. Most respiratory viruses tend to have low infection rates, apparently because they would otherwise quickly wipe out their host, Schaffer said.
Schaffer's technique forced the normally benign AAV, which has already infected over 90 percent of people without any harmful side effects, through rounds of directed evolution to increase its infectivity several hundred-fold.
"We devised a way to evolve viruses that are released from the natural constraints of evolution and have the freedom or ability to evolve toward properties that are more useful for medical application," he said. "In human lung tissue, it completely rescued the chloride ion transport properties of the cells after delivering the correct copy of the CFTR gene to replace the mutated copy of the gene that is present in cystic fibrosis patients."
In this case, the infectious AAV strain developed two major changes: Thanks to a mutation on the viral surface, it was able to bind to different receptors or bind to a more plentiful receptor on the cell surface; and it also acquired a mutation that gave the virus an enhanced ability to make it past the cell surface membrane, slip past the lipid bilayer and reach the inside the cell.
"Neither change alone was enough; it had to be the combination of the two that resulted in the improved properties," said Schaffer. "If we decided to use rational design, we wouldn't have known this. So, we left it up to evolution to discover the answer."
Coauthors with Schaffer and Zabner were James T. Koerber, a graduate student with UC Berkeley's chemical engineering and bioengineering departments and with the Helen Wills Neuroscience Institute; research scientist Katherine J. D. A. Excoffon and David D. Dickey of the University of Iowa; Matthew Murtha and Brian K. Kaspar of The Research Institute at Nationwide Children's Hospital in Columbus, Ohio; and Saf Keshavjee of Toronto General Hospital and the University of Toronto.
The research was funded by the National Institutes of Health and the Cystic Fibrosis Foundation.